过敏性紫癜发病机制,你了解全了吗?
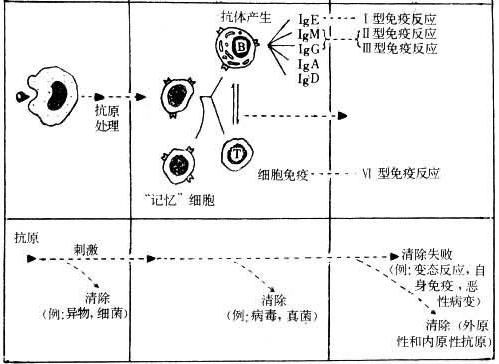
目前过敏性紫癜的发病机制尚不清楚。主要与变态反应体液免疫异常有关。T淋巴细胞功能改变、细胞因子和炎症介质的参与在发病中起重要作用。经过大量实验室病理分析和临床病案的研究,我们发现机体接受外物(感染源、过敏原或其他诱因),引起自身免疫变态反应,生成抗原抗体复合物沉积于血管壁,引起血管壁结构和功能改变,导致皮肤及器官微小动脉和小血管出血性炎症,是主要原因之一。
过敏性紫癜基础病理 由致敏原与体内蛋白质结合,形成抗原。产生的IgE抗体吸附在肥大细胞上,释放出组胺及慢反应物质。这类物质引起小动脉及毛细血管扩张,血管通透性增加,血浆及血细胞渗出,引起水肿及出血。
抗原传递示意图
小动脉及小静脉也可受累,抗原-抗体复合物可刺激嗜碱粒细胞释放组胺及5-羟色胺,也可沉着于血管壁及肾小球的基底膜上激活补体,引起组织损伤。小血管的周围有中性粒细胞、单核细胞、淋巴细胞,也可有嗜酸粒细胞的浸润及不同程度的红细胞渗出,受累血管的周围还可有核的残余及肿胀的结缔组织,小血管的内膜增生,并出现透明变性及坏死,使血管腔变窄,甚至梗塞,并可见坏死性小动脉炎。
皮肤及胃肠道可见坏死性小动脉炎,关节腔内多见浆液及白细胞渗出,但无出血,输尿管、膀胱及尿道粘膜可有出血,并常累及肾脏,紫癜性肾炎的病理变化轻重不等。轻者为局灶性肾炎,重者为增殖性肾炎伴新月型改变,免疫荧光检查可在肾小球上发现C3和IgG,还可见到纤维蛋白原沉积,在血管系膜上也发现有IgA。
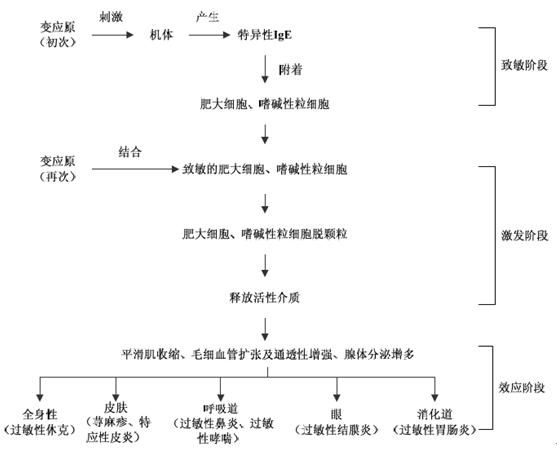
(一)速发型变态反应
致敏原进入机体后,与体内蛋白质结合形成抗原,抗原经过一定的潜伏期(5~20天),刺激免疫组织和浆细胞产生IgE。IgE吸附于全身各个器官的肥大细胞上(血管周围、胃腔、肾小球、皮肤)。当再遇到同一抗原刺激时,抗原便与吸附在肥大细胞上的IgE相结合,激活该细胞中的酶系统,使肥大细胞释放出一系列的生物活性物质,如组胺、5-th、缓激肽、过敏慢反应物质(srs-a),也能兴奋交感神经,释放乙酰胆碱。srs-a是由白三烯c4(ltc4)及其代谢产物lte、ltd4所组成。ltc4在γ谷氨酰转肽酶作用下,转变为ltd4,后者在二肽酶作用下转变为lte4。这一系列生物活性物质,主要作用于平滑肌,引起小动脉、毛细血管扩张、通透性增加,组织、器官出血、水肿。
(二)抗原-抗体复合物反应
这是主要发病机制。致敏原刺激浆细胞产生IgG(也产生IgM和IgA),后者与相应抗原结合形成抗原抗体复合体,其小分子部分属可溶性,在血液中可以沉淀于血管壁或肾小球基底膜上,激活补体系统所产生的c3a、c5a、c5.c6.c7可吸引中性粒细胞,后者吞噬抗原-抗体复合物,释放溶酶体酶,引起血管炎,累及相应器官。另一部分免疫复合物中,抗体多于抗原,复合物分子量大,属非溶性者而沉淀下来,被单核巨噬细胞系统所清除,一般不产生病理变化。
(三)细胞因子的作用
已有报道:过敏性紫癜患者血清中tnfα和可溶性tnf受体(stnfr)在正常范围,而sil-2r水平升高。在伴有肾脏损伤的过敏性紫癜患者肾局部组织细胞中,有多种致炎因子如il-1α、il-1β、tnf-α和lt等的表达。
最近又有报道过敏性紫癜患者尤其是急性期血清中il-4水平明显升高,是正常人水平上限的5~40多倍,提示细胞因子参与过敏性紫癜的发病机制。il-4促进IgE合成,可能是该病过程中的重要因素。
(四)变态反应体液免疫异常
过敏性紫癜患儿体液免疫功能紊乱,B淋巴细胞多克隆活化,患儿血清IgA水平增高,以IgA及IgA免疫复合物沉积于小血管,造成皮肤等血管内皮损伤。欧洲抗风湿病联盟会议已将活检示皮肤或肾小球基底膜上IgA类免疫复合物沉积作为过敏性紫癜的主要诊断标准之一。可见IgA在过敏性紫癜发病中的重要地位。过敏性紫癜患儿血清IgA1水平明显升高,IgA1沉积于小血管壁及由此引起的炎症反应和组织损伤在过敏性紫癜发病过程中起重要作用。IgAl氧连接枢纽区的糖基化异常及IgA1分子清除障碍是导致IgAl免疫复合物(血管毒素)沉积的主要原因。
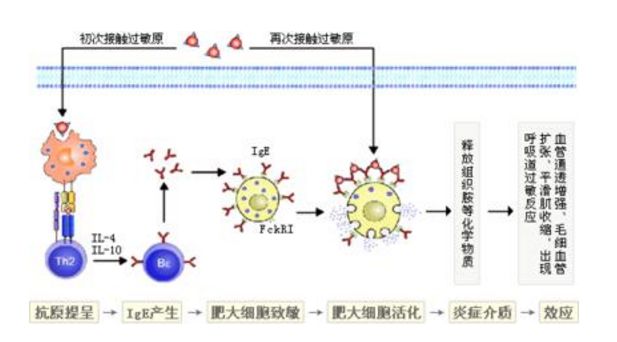
在过敏性紫癜患儿血清中发现循环的IgA型抗中性粒细胞胞质抗体,以及IgA型类风湿因子表达增多,证实IgA免疫复合物在过敏性紫癜发病中可能起关键作用。各种类型的过敏性紫癜均有小分子的IgAl循环免疫复合物(血管毒素)沉积,大分子的IgAl-IgG循环免疫复合物沉积于肾脏是引起紫癜性肾炎的重要原因,而半乳糖缺乏的IgA1水平增高可能在导致紫癜性肾炎发生及判断紫癜性肾炎预后中起关键作用。
(五)T淋巴细胞异常
在过敏性紫癜及紫癜性肾炎发病中,Thl/Th2细胞失衡,TH2细胞过度活化已成为共识。而调节性T淋巴细胞的减少引起免疫抑制效应不足,很有可能是过敏性紫癜急性期免疫失衡的重要原因。
(六)炎症介质
1.细胞因子
IL-6.IL4.TNF-α在过敏性紫癜患儿血清中表达水平显著升高,可能是致过敏性紫癜发病的重要原因。血清、尿液可溶性粘附分子-1 (sICAM-1)和可溶性血管细胞粘附分子-1(sVCAM-1)在过敏性紫癜急性期水平显著高于恢复期和健康对照,提示尿sVCAM-1和sVCAM-1水平高低可能与过敏性紫癜患儿肾脏病变程度有关。
此外,作为前炎症因子家族中的一员,TNF样凋亡弱化因子也被证实可能调控核因子-KB活化,致皮肤微血管内皮损伤,导致过敏性紫癜发病,同时CCL5.CXCL16及CX3CLl等可能诱导HMEC-1 细胞炎症反应,参与过敏性紫癜的发病。
IL一13Rα2和IL一4.IL一6和IL一8和FNF—α可能在小儿AP及紫癜性肾炎中发挥作用。细胞间黏附分子对启动和增强血管的病理免疫反应及随后的组织损伤起关键性作用。Soylemezoglu等研究发现,血浆IcAM一1在急性期时浓度增高。李成荣等研究发现,ALP患儿外周血单核细胞(PBMC)及其培养上清液所诱导的内皮细胞表面多种黏附分子表达明显增强、PBMC与内皮细胞黏附能力明显提高,而且以严重肾脏并发症患儿黏附分子的表达更为显著,提示黏附分子参与介导循环中的免疫效应细胞向血管浸润,从而在AP损伤的病理生理机制中起重要作用。
Topaloglu等研究发现,血管内皮生长因子在疾病急性期明显增高,提示其具有有效趋化性、迁移性、渗透性的因子,在AP形态学及功能学变化中起重要的作用。
白三烯可能在AP的发病机制中起着前炎症和前纤维化的作用,脂氧素4(LXA4)缺乏可能是导致AP患者病情加重的原因。血浆血栓腺烷A2(TXA2)和前列腺环素(PGI2)的不平衡,可能参与儿童紫癜性肾炎肾损害的进展,TXA2和PGI2的平衡有助于肾功能的恢复。
2.一氧化氮(NO)
导致内皮细胞损伤的NO和对血管具有保护作用的硫化氢(H2S)在相互调节中实现自稳态平衡。在过敏性紫癜急性期时,NO的升高程度明显高于H2S,二者调节失衡可能是导致过敏性紫癜发生的原因之一。NO和内皮索-1(ET-1)水平在过敏性紫癜急性期患儿血浆中异常升高,且在紫癜性肾炎及高血压组中更显著,提示NO和ET-1可能与过敏性紫癜,尤其是紫癜性肾炎的发病相关,NO、ET-1的水平可作为临床判定过敏性紫癜病情及预后的重要指标。
3.补体
早期发现过敏性紫癜患者肾毛细血管壁、系膜区有补体C5.C6.C7.C8.C9沉积,而近期则显示C3与过敏性紫癜有关。在过敏性紫癜患儿治疗前研究发现C3明显增高,检测免疫球蛋白及C3对过敏性紫癜的诊断、鉴别诊断、病情监测、预后判断等有一定的指导意义。
4.其他因素
肾小球及间质区A-平滑肌肌动蛋白表达增高可能与过敏性紫癜及紫癜性肾炎的发病及预后不良相关。此外,血栓素B2与前列环素之间平衡失调,导致血小板聚集,凝血因子合成异常增加,激活内源性凝血系统,均参与了过敏性紫癜小血管炎的发生。
(七)凝血机制异常
AP患儿体内存在不同程度的高凝状态,血浆D—D明显增高,肾脏内皮细胞和系膜区有交联纤维蛋白沉积物,检出率为75%,反映血管内凝血、纤溶系统激活,且与内皮及系膜损伤有关。Motoyama等研究发现,血浆CH50、C3.CA水平明显下降。且与疾病严重性无关。链球菌感染后肾小球肾炎与过敏性紫癜肾炎的合并发生率较高,提示链球菌感染后肾小球肾炎与过敏性紫癜肾炎的发病具有相关性。
